Calculating First Derivatives
[Input: recipes/analysis/derivatives/]
The ability to calculate the first (negative) derivatives \(-\frac{\partial E_i}{\partial \vec{R}_j}\) of system wide network predictions \(E_i\) w.r.t. the atom coordinates \(\vec{R}_j\) is of central importance in terms of inferring properties like atomic forces \(\vec{F}\,^i_{\!j}\).
For networks trained on purely ACSF based features, generated by Fortnet, the
calculation of such derivatives can be conveniently enabled in the Analysis
block of the HSD input. The analysis is based on the central finite difference
(please mind the sign flip that was carried out in order to address the minus
sign in front of the gradients):
Internally, Fortnet uses atomic units which is also valid for the \(\Delta\)-parameter with the dimension being length (unit: Bohr).
This chapter should serve as a tutorial guiding you through your first force calculations using Fortnet. As an exemplary dataset, the energy-distance scan of an \(\mathrm{H}_2\) molecule is used. The procedure is split into two major steps:
providing an appropriate input to Fortnet
extracting and analysing the results
Accordingly, you will have learned all the tools to perform, for example, geometry optimizations or molecular dynamics based on Fortnet’s predictions.
Providing the Input
The specification takes place in the Analysis block of the HSD input:
Data {
Dataset = 'fnetdata.hdf5'
NetstatFile = 'fortnet.hdf5'
}
Options {
ReadNetStats = Yes
Mode = 'validate'
}
Analysis {
Forces = FiniteDifferences {
Delta = 1e-02
}
}
The absolute displacement \(\Delta \vec{R}_j\) of the atomic coordinates is
determined by the Delta parameter, which defaults to
\(10^{-2}\,\mathrm{a_0}\) and may be omitted if this value seems acceptable
to you, i.e.:
Analysis {
Forces = FiniteDifferences {}
}
Examining the Output
As usual in validation or prediction mode the relevant output fnetout.hdf5
is written to disk and contains the additional results of the force analysis.
By using the Fnetout class within the Fortformat Python framework,
the atomic forces may be extracted by accessing the corresponding property:
#!/usr/bin/env python3
'''
Application example of the Fortformat package, based on an output
file that contains atomic forces, resulting from finite differences.
'''
import numpy as np
from fortformat import Fnetout
def main():
'''Main driver routine.'''
fnetout = Fnetout('fnetout.hdf5')
forces = fnetout.forces
# print forces of each datapoint, network
# output and atom to illustrate the indexing:
for idata in range(len(forces)):
for iout in range(len(forces[idata])):
for iatom in range(np.shape(forces[idata][iout])[0]):
print(forces[idata][iout][iatom])
if __name__ == '__main__':
main()
Please note that this is definitely not the most elegant way to extract the data but rather an illustration of the underlying indexing.
If the force vectors of the two hydrogen atoms are plotted together with the corresponding geometries, you should obtain something similar to the following animation:
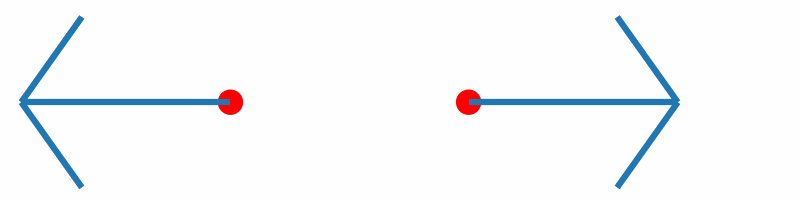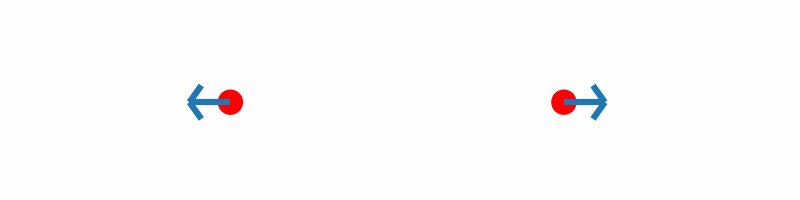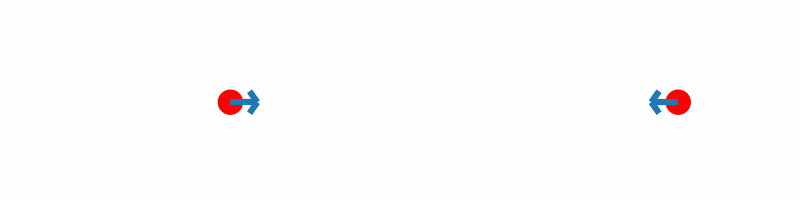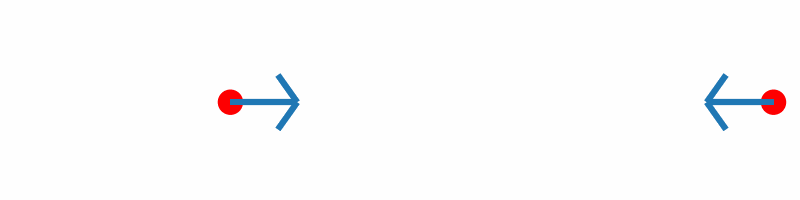
Inline with our expectations, repulsive forces are present for interatomic distances smaller than the equilibrium distance, and attractive forces for distances beyond.